gene editing

Forget AI. Gene editing is still our most powerful — and dangerous — technology.
▸
5 min
—
with

It marks a breakthrough in using gene editing to treat diseases.

In the near-term, gene editing is not likely to be useful. Even in the long-term, it may not be very practical.

Scientists in Japan have genetically modified chickens to lay eggs containing an extremely valuable protein that helps treat cancer, hepatitis and multiple sclerosis in humans. The cost of one of […]

Granted, genetic manipulation has been a dream for decades. Here’s what is different now.

Researchers succeed in deleting key genes from ants, significantly modifying their behavior.
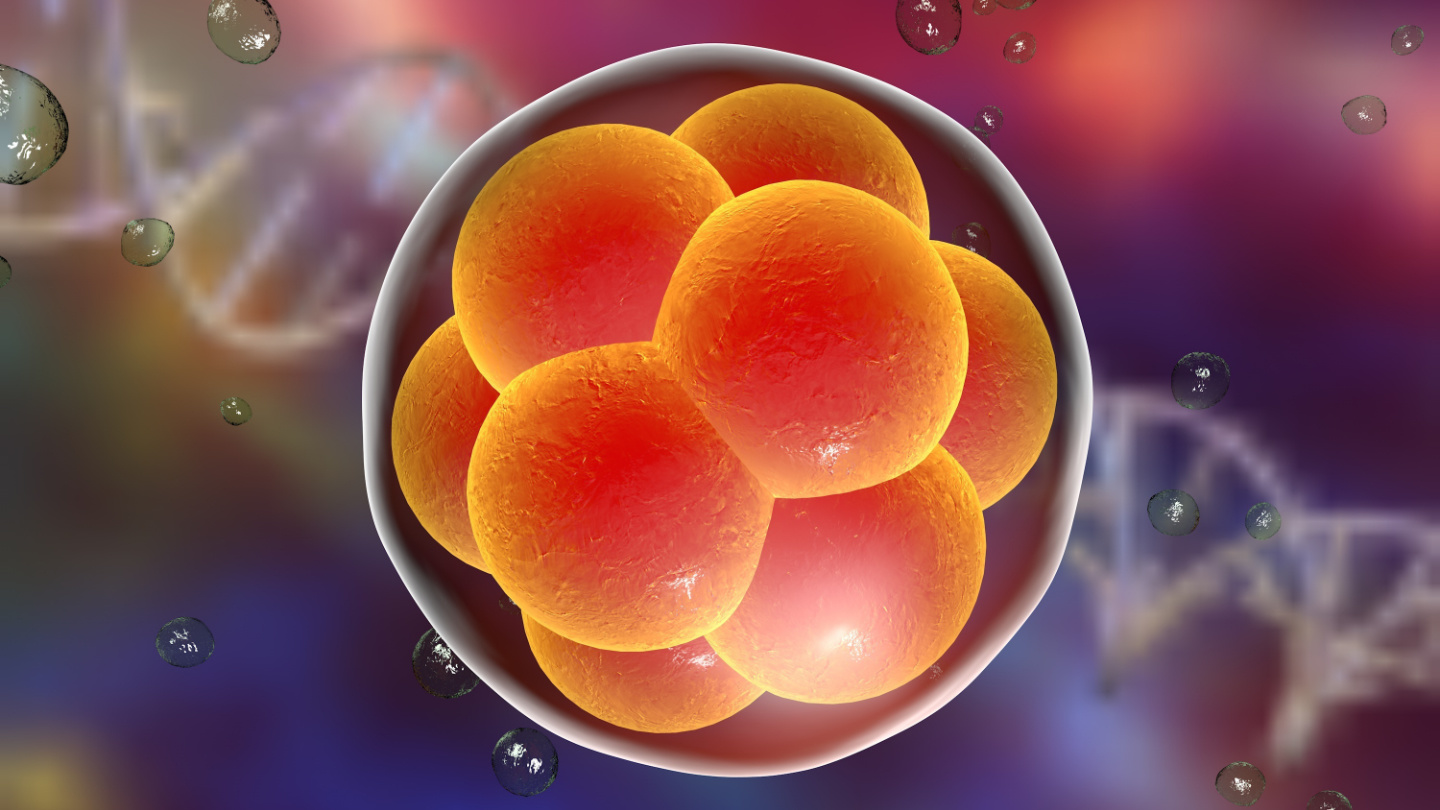
U.S. scientists have successfully repaired DNA in a human embryo for the first time.

Harvard scientists say they are two years away from creating a hybrid embryo with mammoth traits.

Creating a race of super soldiers is off the table, too.
Does life work like our technology? Is life under the hood just like a car sporting souped-up complexity?

A report by a team of scientists highlights the dangers of “gene drive” technology that can eliminate unwanted species.