- Prions don’t sound so bad at first blush: they’re simply proteins that have the wrong shape.
- They may sound innocuous, but “catching” prions is always fatal, and there is no cure.
- Curiously, the most famous case of a prion disease outbreak happened in a cannibalistic tribe in Papua New Guinea.
Until the early 1960s, the Fore people of Papua New Guinea were afflicted with a rare and fatal disease called kuru. When a Forean caught this sickness, they would first become less coordinated and experience uncontrollable trembling and difficulty speaking. Then, the tremble would turn into a shaking. An infected individual would burst out into spontaneous laughter, and they would have difficulty regulating their emotions. In the final stage, the individual would no longer be able to stand or even sit. They would have difficulty swallowing and would fail to react to stimuli. Soon afterwards, they would die.
But the Fore people became free of this disease in 1960s. Since then, the number of new cases of kuru dropped year by year. No vaccine or miracle cure was responsible for this; instead, they simply ceased eating their dead. The Fore practiced funerary cannibalism, eating the bodies of their dead ancestors in the belief that doing so would help free their souls. The brains of the dead were primarily consumed by women and children primarily. For this reason, kuru tended to afflict women and children the most; the brains of the infected dead had the highest concentrations of a wholly unique infectious agent. It wasn’t a bacteria or a virus — it was a simple protein.
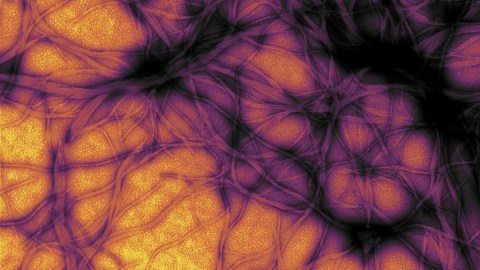
This image depicts a cow’s brain affected by mad cow disease, a kind of prion disease. The small holes are the result of neuronal degeneration due to the accumulation of prions.
Wikimedia Commons
How prions cause disease
Proteins differ according to their shape and composition. One protein made up of certain amino acids and folded into a certain shape will do a certain job in the body. But sometimes these proteins can be folded into the wrong shape, rendering it useless for its intended task.
However, one protein called PrP becomes a bit more problematic when it’s misfolded. We’re not entirely sure what PrP does when it’s working properly — it seems to have something to do with memory and neuron formation. But whenever a PrP protein is made in the wrong shape, it turns into a proteinaceous infectious particle, or a prion. These prions were the infectious agents hiding in the brains of the Fore people’s dead ancestors and are ultimately the cause of their deadly disease.
A single prion wouldn’t be much of a problem in the human body. One instance of a misfolded protein that couldn’t do its job wouldn’t cause serious harm. The trouble is, prions don’t just become inactive when they’re misfolded; they also gain a new capability. The best analogy is that they become zombie proteins — when they come into contact with another PrP protein, they cause that protein to misfold into another prion. This sparks a chain reaction: each prion makes more and more prions. As they accumulate in the brain, they interfere with cellular function and cause neurons to degenerate and die.
Scientists refer to this condition as prion disease, or transmissible spongiform encephalopathy (TSE) due to the sponge-like appearance of the affected brains. As the neuronal degeneration progresses, one can find small holes dotting a diseased brain where the cells have died off. Kuru is one type of prion disease, but this class of maladies also includes mad cow disease, fatal familial insomnia, and Creutzfeldt-Jakob disease.

This image depicts a cow’s brain affected by mad cow disease, a kind of prion disease. The small holes are the result of neuronal degeneration due to the accumulation of prions.
Wikimedia Commons
How one catches a prion disease
There are three ways one can “catch” a prion disease. First, the PRNP gene (which produces the PrP protein) can be mutated, making it more likely for the associated protein to fold into a prion. Prions can form spontaneously as well, though this is fairly rare. Finally, one can ingest prions, as is the case with kuru or mad cow disease. Researchers believe that one individual in the Fore people likely developed Creutzfeldt-Jakob disease spontaneously. Then, when this individual’s relatives ate his or her brain, they also ingested the prions that led to his or her death.
Can prion diseases be treated?
There are two major challenges to dealing with prion diseases: detecting the disease and treating it. Prion diseases are extremely difficult to detect except by examining the brain after death, and they can take decades before they start manifesting symptoms. During this time, however, the disease is still progressing, converting healthy PrP proteins to prions. Unfortunately, these prions don’t accumulate in easily accessible parts of the body, like the circulatory system, making it extremely challenging to test for them.
Prion diseases are always fatal, and there is no cure for them to date. Since prion diseases occur in the brain, any drug that targets prions has to be small enough to pass through the blood-brain barrier, which significantly limits our options for treatment. Still, some progress has been made, the most recent of which examined the use of a compound called SGI-1027, which binds to healthy PrP proteins, preventing them from being zombified by prions. However, this compound hasn’t been tested in living subjects, and other possible treatments are still awaiting human trials.
Gene therapy has so far proven to be a promising method, but this is also a long way away from becoming a reality for most patients. One promising finding was the discovery that some humans possess a genetic mutation that makes them either strongly resistant to or completely immune to prions; they produce slight variations of PrP that simply don’t misfold when they come into contact with prions. This mutation is relatively common in the Fore people, who were more likely to survive if they possessed it. Researchers have introduced this gene into mice, who became correspondingly immune against various prion diseases.
It may also be possible to “turn off” the gene responsible for producing PrP in humans. In this case, even if a patient has already contracted a prion disease, their bodies simple won’t produce any more PrP to be turned into prions, thus preventing the catastrophic buildup of protein that causes cellular damage.
While prion diseases are among the scariest and most unique illnesses out there, it’s important to remember that they are extremely rare. Creutzfeldt-Jakob disease, for instance, only occurs in about one out of every million people. Additionally, it’s possible to contract a prion disease from diseased meat as well. Some may recall the panic spread by mad cow disease; however, one is only likely to contract a prion disease in this way if the animal had a prion disease itself and if the meat had been contaminated by the brain or spinal cord of the animal. And of course, cannibals are at high risk for prion disease, but not many tears would be shed for them anyhow.