What if we tolerated diseases?
When people think about infectious diseases — as many have, these last three years — they think mainly about the immune system. The severity of an individual’s illness, it’s assumed, is down to how well the immune system detects, attacks and eliminates the pathogenic invader.
The immune system is said to resist disease. Resistance reduces the amount of pathogen residing inside a host, thereby curtailing disease progression, driving recovery or preventing infections altogether. People who are immunocompromised fear infections because they cannot effectively resist pathogens.
And vaccines work because they teach immune systems to recognize — and so more effectively resist — pathogens before the actual bug is encountered.
But there have always been nagging issues with so straightforwardly relating illness to the abundance of pathogen in a host. And when Janelle Ayres, a physiologist now working at the Salk Institute in San Diego, entered grad school 20 years ago, these anomalies bothered her. “I was very interested in the conventional thought or assumption that all that’s necessary to survive an infection is that you have to kill the pathogen,” she says. “I was interested in this because there are clearly examples in humans and animal models where this is just too simplistic.”
Most obviously, there are asymptomatic infections. Long before we knew that certain individuals infected with SARS-CoV-2 develop no symptoms, there was the famous case of New York cook Mary Mallon. Christened “Typhoid Mary,” Mallon unknowingly lived with a Salmonella typhi infection in the early 1900s and, in good health, transmitted the bacteria to dozens of people, some of whom died.
Conversely, there are overreactions to an infection — for example in sepsis, where the immune response is dramatically disproportionate to the infection and the whole-body inflammation that’s set off gravely damages host tissues. Often, the causative pathogen has been virtually eliminated but the overactive and maladaptive immune response to it continues to wreak potentially deadly havoc.
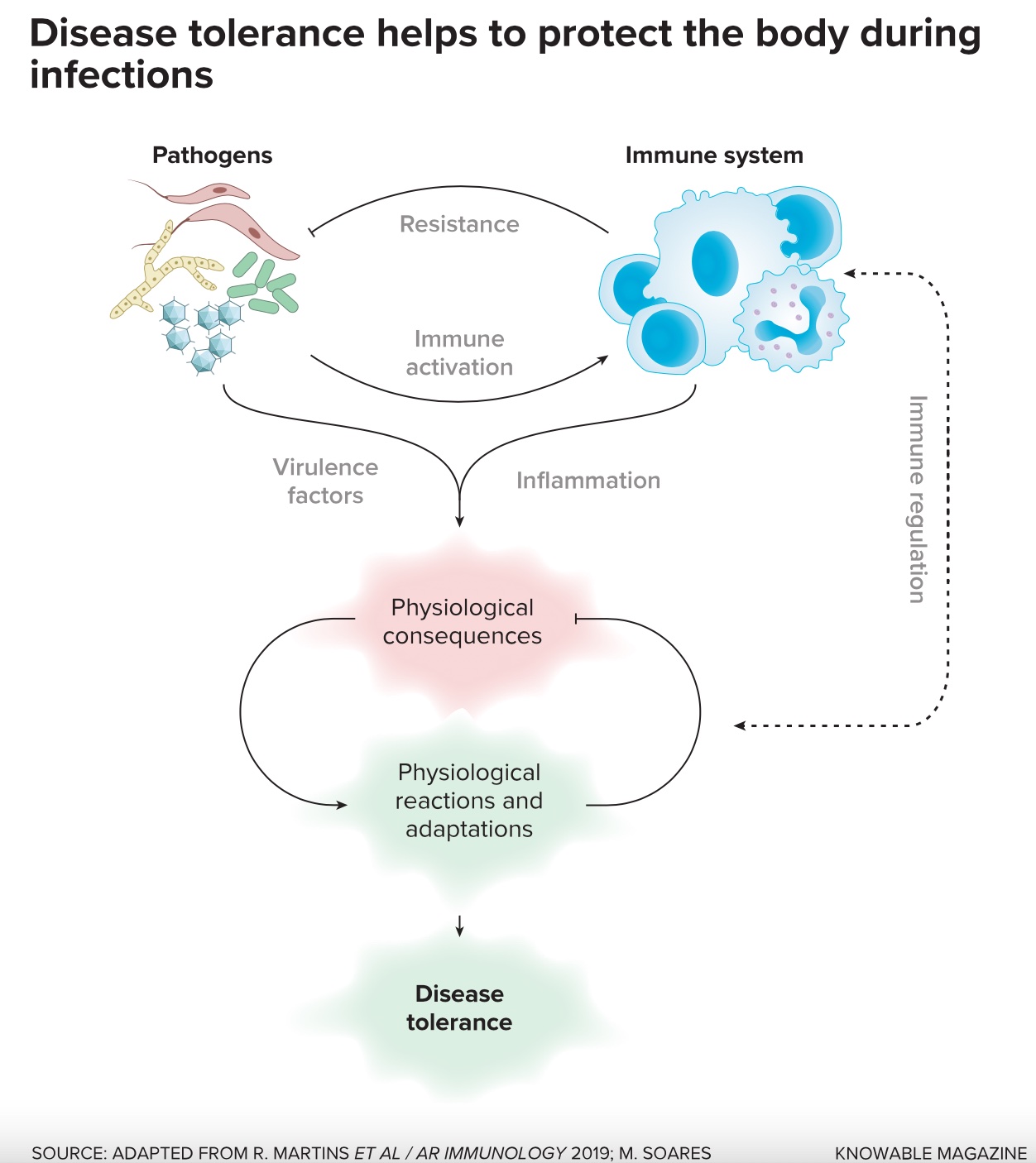
In David Schneider, Ayres found a PhD supervisor who was an expert in infectious disease but keen to move beyond just studying immunology. He wanted to consider the ways that animals interact with microbes growing in and on them, be these harmless, beneficial or disease-causing. Together, the pair helped to kick-start interest in a defense mechanism that, today, they think is central to health and resilience: the ability not to resist an infection but to tolerate one.
Schneider, of Stanford University, had already helped to establish fruit flies and their incredibly well-characterized genetics as a model for studying infectious diseases. But he had started to think that people using this system had become too focused on using it solely to identify new genes involved in the immune response. “I tried to ask a more general question,” he says, “where I said, ‘What does it take for the fly to survive when you give it an infection?’”
To tackle this question, for her PhD Ayres took more than 1,200 fly strains — each with a different genetic mutation — and infected them with the pathogen Listeria monocytogenes. Then she recorded how long the flies took to die. Eighteen strains died more quickly than average; Ayres then measured how many bacteria had grown in each of the more susceptible strains.
In 12 of them, she found that bacterial levels were higher than normal — indicating that in these flies, the mutations had compromised their immune systems and allowed the bacteria to multiply out of control. In the remaining six strains, however, Listeria levels were normal.
These mutant flies were opposites of Typhoid Mary. Whereas her body had been able to handle an infection without any signs of sickness, these were being killed by levels of pathogen that should not have been fatal. Their immune responses were normal, but they appeared to lack some sort of resilience trait that ordinarily allowed flies to survive this infection.
It was an experiment that set the direction of Ayres’s career — for, she says, “it suggested that there are genes that an animal host has evolved that’re important for tolerating an infection.”
The nature of these systems, how they work and how they’re controlled are questions that have occupied Ayres, Schneider and a small but growing number of researchers ever since. “It’s easy to describe what a diseased individual is. We know the mechanisms that lead to disease,” says Ayres. “But how can you have an individual that … has a pathogen, but they’re healthy? How do you describe that mechanistically? What’s going on?”
Today, there is a substantial body of research showing various ways in which animals can tolerate, and so survive, maladies such as malaria, sepsis and dysentery. And while there is a feeling among the scientists working in this field that their findings have not yet influenced work on infectious disease as much as they’d like, they are optimistic that their research could help to forge important new medical therapies. “Hopefully we’re finding core mechanisms that protect us against infectious diseases,” says Miguel Soares, an immunologist at the Gulbenkian Science Institute in Portugal, who coauthored an article about disease tolerance in the 2019 Annual Review of Immunology.
In fact, Soares hopes that basic research has already identified one such way. A drug that allows mice to survive sepsis by engaging the animals’ intrinsic resilience mechanisms is now in the first human clinical trial born directly of disease tolerance research.
Replanting an old idea
Schneider and Ayres did not invent the concept of disease tolerance. In fact, it was already more than a century old. As a grad student, Ayres read the work of a peripatetic American scientist named Nathaniel Augustus Cobb. In the late 19th century, working for the newly founded Department of Agriculture in New South Wales, Australia, Cobb had attempted to boost the productivity of local farms. In doing so, he described certain strains of wheat that developed robust fungal infections yet kept on growing and cropping — the pathogen was there but it was barely diminishing these plants’ vitality.
Through the 20th century, plant scientists embraced and dissected this defense mechanism, but the idea never caught on with scientists studying animals. Animals, after all, have immune systems — and that’s what animal and clinical research focused on.
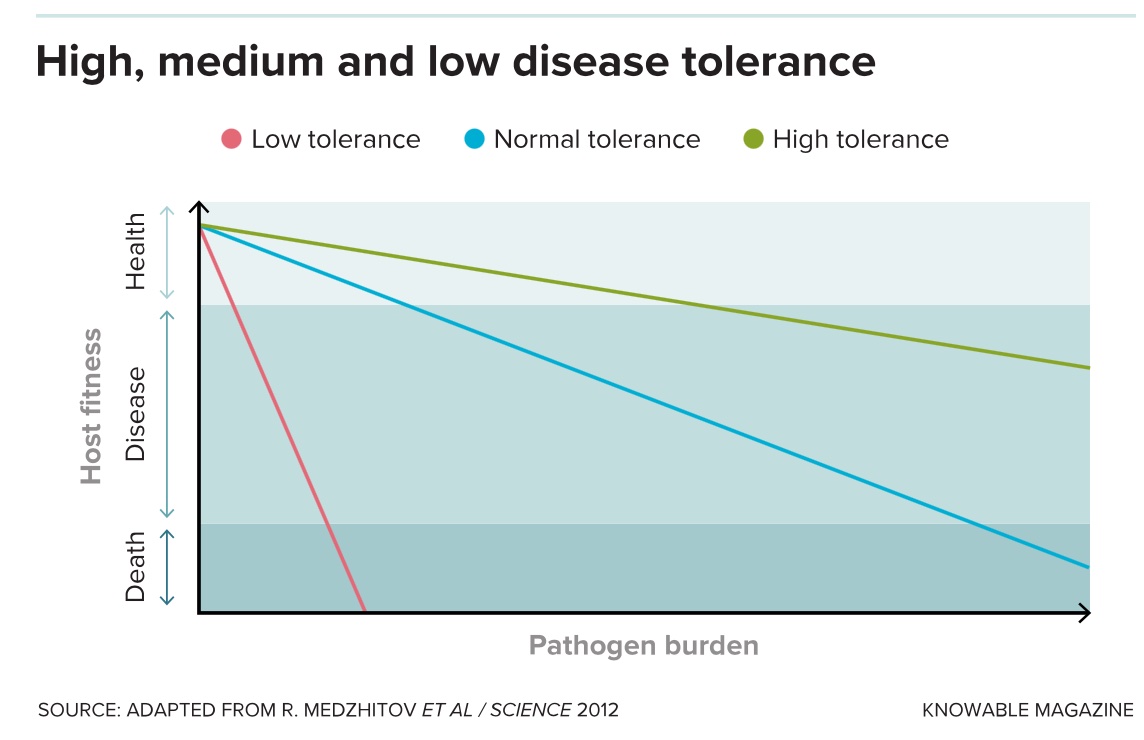
Then, just as Schneider and Ayres were exploring this concept in fruit flies, a group at the University of Edinburgh in Scotland published on similar work in mammals. The study examined what happened when several strains of mice were infected with the malaria-causing parasite Plasmodium. Some mouse strains had tolerated high levels of the parasite before becoming sick, while in others, low levels had caused illness. The authors concluded that in mice, pathogen levels alone could not explain disease severity — meaning mammals, too, vary according to how well they tolerate a pathogen, and this variability is genetically controlled.
These twin demonstrations that disease tolerance applies to animals as well as plants was a “very simple kind of conclusion, but very deep,” Soares says. “That’s when we all woke up!”
For one thing, the work suddenly offered a new framework in which to reinterpret sometimes puzzling old data.
Soares mainly studies malaria and sepsis — diseases that, respectively, kill around 600,000 and 11 million people annually. He had previously found that when mice were infected with the parasite that causes malaria, the activity of a gene critical for metabolizing iron increased. This allowed the mice to better handle the heightened levels of free iron in their bloodstreams brought about by the infection, thereby improving survival rates.
Suddenly, this seemed like an example of tolerance. And this sort of detailed mechanistic understanding was exactly what was needed to move from an idea (animals have non-immune-cell-mediated defenses against damage wrought by pathogens that have colonized their bodies) to a mature field of biology (this is how animals tolerate pathogens).
Soares stresses that advocates of disease tolerance are not downplaying the centrality of immunity. Rather, they propose that these two protective systems work together. He is also clear in his belief that vaccination and antimicrobial drugs remain the most powerful ways to combat infectious diseases. “But in some cases,” Soares says, “that’s not enough.” What he wants are additional ways of restricting the illness that an infection causes.
Ayres, Soares, Schneider and others have now looked at multiple pathogens and interrogated how animal bodies respond to them. They have found various means by which health is maintained — or is not. These mechanisms are often complex, but that shouldn’t be a surprise, they say: The direct damage caused by pathogens induces an array of physiological reactions that, in turn, have numerous knock-on effects. And all of these can matter. Frequently, the biology of the whole animal is involved, not just the organ systems that the invader attacks. Progress is being made in mapping these multifaceted pathways.
Sweet success
Initially, Soares’s group focused extensively on iron in their studies of both malaria and sepsis. Each disease can cause the breakdown of iron-containing red blood cells, which releases iron-containing heme proteins that need to be dealt with in order for the body to survive. But there is a catch. Neutralizing the proteins liberates free iron, which is toxic to the host and is also a mineral that bacteria and other parasites need to survive. So dealing with these iron-containing proteins involves a fine balance between tolerating and succumbing to disease.
In a 2017 paper, Soares and colleagues found two treatments that allowed mice to better survive sepsis. The first was to treat them with a protein that safely mops up iron. The second was to give them glucose.
“Bacteria, they like glucose,” says Soares. Therefore, the body often shuts down glucose production when it is infected, and infections often make animals lose their appetites. These twin actions limit the pathogen’s energy source and so help survival of the host.
In their sepsis model, Soares’s lab found that the changes to iron metabolism had caused glucose production by the liver to decrease — but that this could go too far. “We found that you can drop the glucose, but you cannot drop it below a certain threshold level. Below that threshold level, you develop hypoglycemia and you die,” says Soares. When Sebastian Weis, a postdoc working in his lab, gave dying mice sugar, the animals rallied and lived.
Such an intervention does not help a host eliminate the disease-causing bacteria; rather, it allows it to carry on functioning until that pathogen is gone. It is, in other words, fostering disease tolerance.
Work elsewhere has reported that maintaining blood glucose in a functional range is also key to tolerating other infections; the precise responses are likely to be specific to the pathogen in question, Soares says. Ayres, meanwhile, has demonstrated just how rich and dynamic the relationship between host and a parasite can be — and how glucose levels can be key to this interaction.
To begin studying a new disease, Ayres employs a clever experimental system that entails giving a group of genetically identical mice a dose of pathogen that she knows will kill half of them. She then compares the reactions of survivors and non-survivors to ask what made the critical difference between life and death.
When she decided to use this approach to investigate a strain of Citrobacter — a mouse pathogen that shares virulence factors with E. coli strains that can cause disease in people — “I thought for sure all of the animals would get sick, and then some would recover,” she says.
But that’s not what happened. The mice that lived never got ill. Whatever was saving them kept them healthy all along.
As always with an experiment of this type, Ayres’s team checked whether the surviving mice had simply mounted a quicker, more effective immune response. But they found the amount of Citrobacter in the animals that had died and the ones that had survived were the same. Their immune responses were equivalent.
Turning to the animals’ metabolisms, Ayres’s team saw that surviving animals had changed the way they metabolized iron, and that this, in turn, had made them produce more free glucose in their circulation.
To explore the consequences of this, Ayres asked what effect the raised sugar levels had on the Citrobacter. “It’s really important when you’re studying host-pathogen interactions — or any system where you have two entities interacting — to consider both,” she says. It turned out that when these particular bacteria had their energy demands met by the host, they turned off their disease-causing virulence mechanisms. The bacteria had ceased to be pathogenic invaders and become, instead, essentially harmless microbes coexisting with their healthy host.
But while this suggested a way to protect individuals infected with Citrobacter, the result raised a troubling possibility. What if the surviving mice became walking reservoirs of lethal bacteria, basically little murine Typhoid Marys capable of spreading disease?
Instead, Ayres found something quite different. Citrobacter in tolerant mice quickly became less virulent. And when the bacteria from tolerant mice were transferred to uninfected mice, they didn’t cause illness. In other words, it looked as though well-fed bacteria happily multiplying in their hosts had quickly adapted to live that way, chronically dialing down their virulence. Ayres terms the way that changes in sugar and iron regulate Citrobacter a form of “metabolic bribery.”
“I thought it was terrific — this concept,” says Elina Zuniga, an immunologist at the University of California, San Diego, who was not involved in the work. Zuniga says it highlights how, when the immune system suppresses pathogens, it puts the microbe in a corner, pressuring it to evolve more virulence. In tolerating an infection, Zuniga says, “we’re avoiding this endless arms race between the pathogen and the host.”
From a scientific perspective, these studies give important insights into disease biology and how the trajectory of an infection is shaped. But in terms of their clinical usefulness, Soares’s colleague Weis — who now splits his time between basic research and working as an infectious disease specialist at Jena University Hospital in Germany — is blunt. “The glucose story is boring for clinicians,” he says, and in many ways, managing hypo- and hyperglycemia is already clinical common sense.

What clinicians want, Weis says, are new drugs — and he is leading the first clinical trial that might introduce one, for sepsis.
Switching on defenses
Weis’s own research with Soares suggested that people with sepsis might be helped, as mice were, by proteins that mop up free iron. But since he knew of no such protein available for clinical testing, he took an alternative approach.
He focused instead on what he calls the tissue damage-control response — another mechanism that he believes is elemental to disease tolerance.
Soares’s lab has found that a gene called Nrf2 is switched on in cells undergoing some form of stress and that this gene helps to protect the cells. Nrf2 controls a whole network of genes that serve a damage-control function, and this network appears to be activated whenever tissues are stressed.
Soares first observed this tissue damage-control response in mice with malaria. What’s critical about it, he says, is that “it doesn’t touch the pathogen. It just protects you from the damage.” That includes damage caused directly by pathogens, as well as the inevitable collateral damage mediated by an activated immune system and inflammation. This collateral injury is what makes sepsis so dangerous.
The research suggested that if there was a way of switching on the tissue damage-control system, it might quell disease severity. And it appeared that there was such a way. In 2013, a Portuguese group that Soares knows well showed that mice with sepsis could be saved by giving them epirubicin — a 40-year-old drug used in chemotherapy. Epirubicin induces DNA damage, which at high doses kills cancer cells. But at lower doses, it appears to disrupt things just enough to switch on the tissue damage-control response, thereby protecting the mice and preventing sepsis from killing them.
“The way we perceive this,” says Weis, “is if you have an infection and your system is able to adapt to the infection-associated stress, you will not get organ failure.”
Weis is now starting a clinical trial to see if this holds true for humans. The big plus of using epirubicin is that it is already used in humans. The downside is that regulators told Weis he must begin with very low doses to ensure it is safe in sepsis patients. He thinks that within the next year he will have safety data from roughly 45 patients, plus data indicating whether epirubicin does, indeed, induce tissue damage-control mechanisms in people.
All being well, Weis will then test whether the drug improves sepsis outcomes.
Researchers in this field will keep their antennas up for other ways to exploit the science clinically, even as they continue to press the fundamental biology forward. Ayres, for example, wrote early in the pandemic about the need to find ways of protecting people with Covid-19 from the damage caused by their own immune responses; anti-inflammatory drugs such as dexamethasone have, indeed, been life-saving. And she is now bidding to incorporate disease tolerance mechanisms into a new view of what she calls the biology of physiological health.
Ayres also ponders what’s going on in her experimental setup where genetically identical mice have a 50-50 shot at surviving various kinds of infections. What tips the balance one way or the other? “Is it pre-existing? Or is it something that happens in the infection before they’re symptomatic?” she asks. There are multiple things to look at, she adds, from the bacteria that grow in the animals’ guts — their microbiomes — through to their social interactions, individual metabolic differences or maybe even when they last ate. “We really want to know,” she says.
This article originally appeared in Knowable Magazine, a nonprofit publication dedicated to making scientific knowledge accessible to all. Sign up for Knowable Magazine’s newsletter.
![]()





