A revolutionary new CRISPR treatment for sickle cell anemia may be imminent
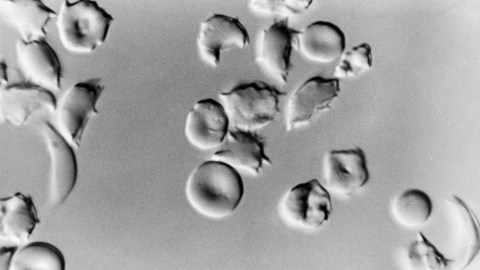
- Sickle cell disease (SCD) is a genetic disorder that affects red blood cells, causing them to become rigid and "sickle" shaped, leading to serious and debilitating health complications.
- Exa-cel, a new gene therapy treatment using CRISPR technology, shows promise in treating SCD by editing the patient's stem cells to produce fetal hemoglobin.
- While the FDA advisory committee acknowledges the potential risks of off-target genetic changes from the therapy, the overwhelming benefit observed in the trials suggests that Exa-cel is likely to be approved.
I have a long-standing interest in sickle cell anemia, a genetic abnormality that is the scourge of approximately 100,000 Americans, primarily Black, who are afflicted with it.
Sickle cell disease (SCD) is an inherited disorder marked by abnormal hemoglobin, the protein that delivers oxygen to the cells of the body. Normal red blood cells are disc-shaped and flexible enough to move smoothly through the blood vessels. In SCD, red blood cells become crescent or “sickle” shaped due to a genetic mutation in the patient’s hemoglobin.
These misshapen red blood cells are inflexible and get stuck in blood vessels, and the resulting impaired blood flow can lead to a variety of complications, including stroke, infection, episodes of pain called “pain crises,” and arthritis from hemorrhaging into joints.
The cause of sickle cell disease
I first became aware of SCD during the 1960s because my MIT undergraduate advisor, Professor Vernon Ingram, had unraveled its molecular basis: a mutation in DNA that causes a change in a single amino acid in the hemoglobin molecule. (Proteins are comprised of chains of amino acids.) The change occurs specifically at one site where normal hemoglobin has an amino acid called glutamic acid; those with SCD have valine, instead. This was the first description of the molecular abnormality in a genetic disease. As a medical resident a few years later, I had a patient with SCD who, by the age of 20, had suffered from most of the awful complications I already mentioned.
Currently, a bone marrow transplant is the only treatment for some patients with SCD, but a new, potentially revolutionary approach might soon be available. The FDA is nearing the end of its evaluation of Exa-cel, a CRISPR-based gene therapy approach to reversing the genetic defect. The treatment involves gene editing of the patient’s blood-forming stem cells to induce them to produce high levels of fetal hemoglobin (HbF, or hemoglobin F) in red blood cells. HbF is the form of the oxygen-carrying hemoglobin that is naturally present during fetal development, but the body switches to the adult form of hemoglobin after birth. The increased production of HbF by Exa-cel reduces painful and debilitating sickle crises for patients with SCD.
Exa-cel clinical trials
On October 31, the FDA’s Cellular, Tissue, and Gene Therapies Advisory Committee discussed Exa-cel. Interestingly, its efficacy and short-term safety were not at issue because the FDA felt that they had been amply demonstrated during the clinical trials.
The demonstration of efficacy was impressive. In the pivotal clinical study, 29 of 30 evaluable patients (96.7%) achieved the primary endpoint of the absence of severe blood vessel blockage (known as vaso-occlusive crises, or VOCs) for at least 12 consecutive months. Of those 29 patients, 28 remained free of VOCs for an average of 22.3 months, with a maximum of 45.5 months.
In addition, all 30 evaluable subjects met the important secondary endpoint of freedom from inpatient hospitalization for severe VOCs for at least 12 consecutive months. My patient decades earlier had had about 80 hospital admissions by the age of 20.
The FDA advisory committee was charged with discussing the likelihood of hypothetical “off-target” — that is, incidental and unwanted — genetic changes caused by the therapy. According to accounts of the meeting, there seemed to be a consensus that the risks were uncertain but probably small. A guest speaker, Dr. Daniel Bauer, director of the gene therapy program at Boston Children’s Hospital and the Dana-Farber Cancer Institute, put it this way:
“Only a small part of the human genome actually codes for genes — most of the human genome is non-coding. It’s likely that many places in the human genome can tolerate an off-target edit and not have a functional consequence. The challenge is that we don’t really know for sure, and the only way to know that is careful follow-up. My guess is that it’s a relatively small risk in the scheme of this risk-benefit, but it’s new, it’s unknown.”
By the end of the meeting, it appeared that most members of the committee believed that the benefits of the therapy far outweigh the theoretical risks of off-target editing. My guess is that the FDA will approve Exa-cel with the understanding that there will be long-term follow-up and collection of data, whether via a Phase 4 (post-marketing) study or simply following the treated patients.
Professor Ingram, who became known as “the father of molecular medicine,” would be pleased.